
When two pure substances mix under normal conditions there is usually an increase in the entropy of the system. This is qualitatively easily visualised in terms of the increased disorder brought about by mixing. For the mixing of two ideal gases it is straightforward to obtain an expression for the entropy of mixing. Since the molecules of ideal gases do not interact the increase in entropy must simply result from the extra volume available to each gas on mixing. Thus, for gas A the available volume has increased from VA to (VA + VB). By calculating the entropy of expansion of each gas we can calculate the entropy of mixing as shown in the panel below.

Experimentally, equation (E2) is found to account surprisingly well for the entropy of mixing for molecules that only interact via van der Waals forces, even when the sizes of the two species differ by factors of two or three. This indicates that the two assumptions made in the derivation above largely cancel each other. This does not occur when the differences are very large, as in polymer solutions (see Third Year Lecture Course on Amphiphiles and Polymers), and it would also not be wise to scrutinize too hard the details of the derivation above. It has only been given to provide a justification of equation (E1). There are other ways of justifying equation (E1) based on lattice models, which have different merits and different drawbacks. Although equation (E1) cannot in any sense be proved, it is one of the cornerstones of models of mixing of liquids.
We now give a possible definition of an ideal solution. It is one for which the entropy of mixing is given by equation (E2) and the enthalpy of mixing is zero. This immediately leads to the result that the Gibbs free energy of mixing is
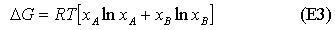
Since the mole fractions in equation (E2) are always less than unity, the ln terms are always negative, and the entropy of mixing is always positive. Its variation with concentration is shown in the diagram below. For the ideal solution, as defined above, the Gibbs free energy from equation (E3) is always negative and becomes more negative as the temperature is increased (see diagram). The decrease in free energy on mixing is always a strong force promoting solubility.
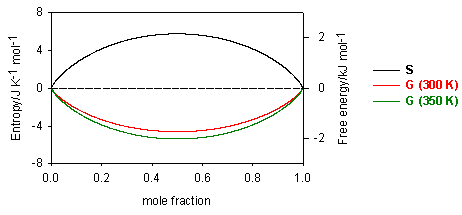
It is interesting to use equation (E3) to derive the relation between DG and the equilibrium constant K. This is not a standard procedure and is not useful except that it does demonstrate very clearly the role of mixing in helping to determine the position of chemical equilibria.
{kind=link}